Introduction to the Data in PRECISION.seq
Intro_Data.RmdMicroRNA (miRNA) sequencing data were collected for two subtypes of soft tissue sarcoma: myxofibrosarcoma (MXF) and pleomorphic malignant fibrous histiocytoma (PMFH). 27 MXF samples and 27 PMFH samples were collected from newly diagnosed untreated tumors at Memorial Sloan Kettering Cancer Center between 2002 and 2012. These tumor samples were sequenced twice: once with uniform handling and balanced sample-to-library-assignment to minimize data artifacts due to experimental handling and avoid their confounding with sample group (that is, tumor subtype); and a second time without, which resulted in excessive artifacts due to handling. The first dataset can be used to assess microRNAs’ differential expression status, serving as a benchmark; the second dataset can be used to test normalization methods against the benchmark. These two datsets are named as data.benchmark and data.test in this package.
We show (1) how to examine the overall distribution of each dataset, (2) how to perform differential expression analysis for each data set and compare the results between the two, and (3) how to create additional paired data sets based on a novel data permutation algorithm and analyze them.
Benchmark Data Analysis
Overall Data Distribution
We examine the overall distribution of the benchmark data (as log2 transformed read counts) using sample-specific boxplots.
benchmark.log <- log2(data.benchmark + 1)
boxplot(benchmark.log,
col = ifelse(grepl("MXF", colnames(benchmark.log)),
rainbow(2)[1], rainbow(2)[2]),
ylab = "Log2 Count", ylim = c(0, 20),
xaxt = "n", outline = FALSE)
legend("topright",c("MXF", "PMFH"), bty = "n",
pch = "x", cex = 1, col = c(rainbow(2)[1], rainbow(2)[2]))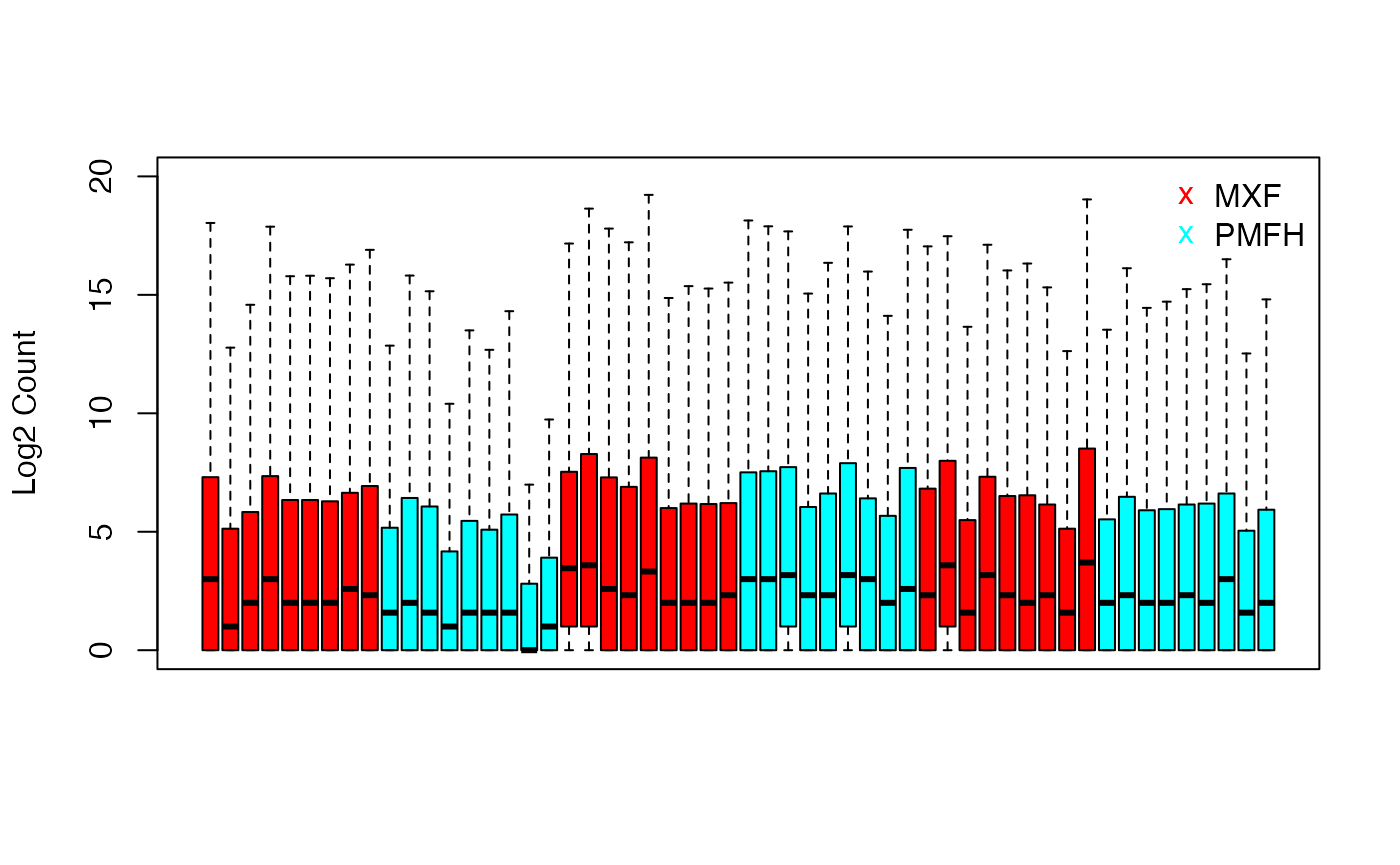
Differential Expression Analysis
We assess evidence for differential expression in the benchmark data using the voom-limma method. The analysis results are displayed with the volcano plot and the scatter plot of group mean difference versus group mean average. Red dots indicate the microRNAs that are significantly differentially expressed (\(p < 0.01\)).
benchmark.voom <- DE.voom(RC = data.benchmark, groups = data.group, Pval = 0.01)
DE.bench <- benchmark.voom$id.list
benchmark.voom.dat <- data.frame(dm = benchmark.voom$log2.FC,
p.value = benchmark.voom$p.val)
mask <- with(benchmark.voom.dat, p.value < .01)
cols <- ifelse(mask,"red", "black")
with(benchmark.voom.dat, plot(dm, -log10(p.value), cex = .5, pch = 16,
col = cols, xlim = c(-3.6, 3.6),
ylim = c(0, 6),
xlab = "Mean Difference: PMFH - MXF"))
abline(h = 2, lty = 2)
cols <- ifelse(rownames(benchmark.log) %in% DE.bench, "red", "black")
plot(rowMeans(benchmark.log),
apply(benchmark.log[,grepl("MXF", colnames(benchmark.log))], 1, mean) -
apply(benchmark.log[,grepl("PMFH", colnames(benchmark.log))], 1, mean),
pch = 16, cex = 0.5, col = cols,
xlab = "Group Mean Average of Log2 Count",
ylab = "Group Mean Difference of Log2 Count",
main = "")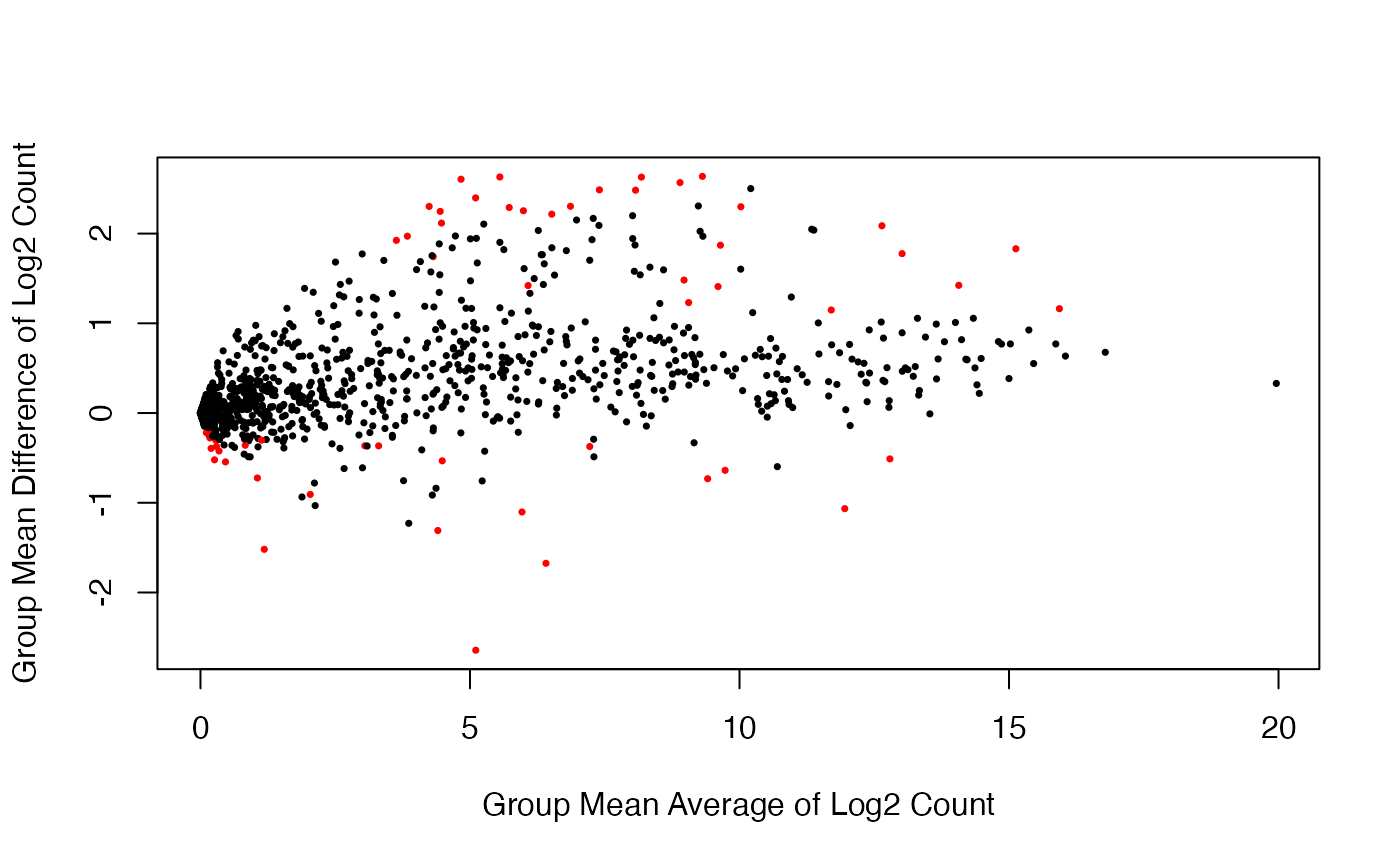
Test Data Analysis
Overall Data Distribution
We examine the overall distribution of the test data (as log2 transformed read counts) using sample-specific boxplots.
test.log <- log2(data.test + 1)
boxplot(test.log,
col = ifelse(grepl("MXF", colnames(benchmark.log)),
rainbow(2)[1], rainbow(2)[2]),
xaxt = "n", outline = FALSE, ylim = c(0, 20))
legend("topleft",c("MXF", "PMFH"), bty = "n",
pch = "x", cex = 1, col = c(rainbow(2)[1], rainbow(2)[2]))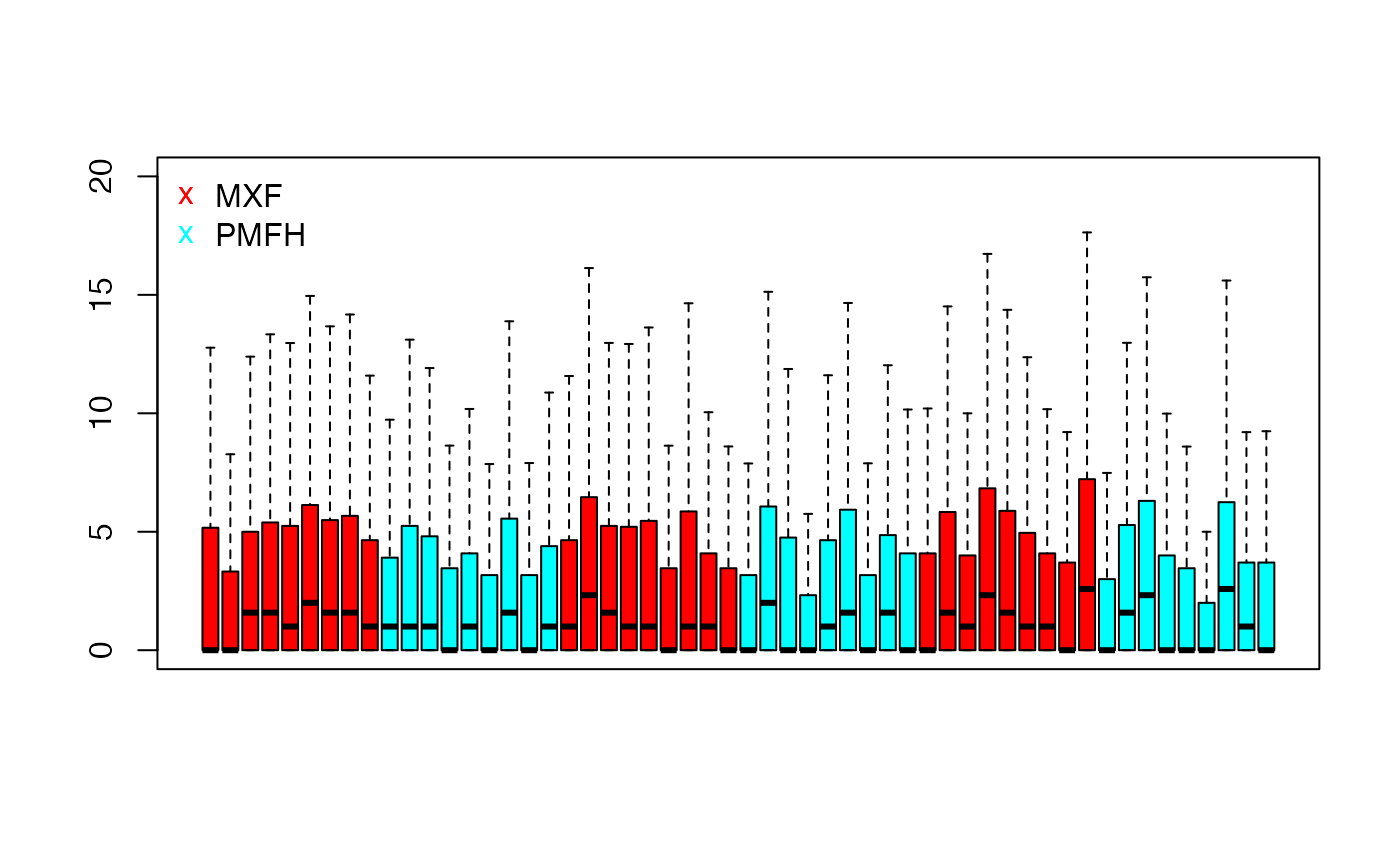
Differential Expression Analysis
We assess evidence for differential expression in the test data using the voom-limma method (without depth normalization). The analysis results are displayed with the volcano plot and the scatter plot of group mean difference versus group mean average. Red dots indicate the microRNAs that are significantly differentially expressed (\(p < 0.01\)).
test.voom <- DE.voom(RC = data.test, groups = data.group, Pval = 0.01)
test.voom.dat <- data.frame(dm = test.voom$log2.FC,
p.value = test.voom$p.val)
mask <- with(test.voom.dat, p.value < .01)
cols <- ifelse(mask,"red", "black")
with(test.voom.dat, plot(dm, -log10(p.value), cex = .5, pch = 16,
ylim = c(0, 6), xlim = c(-3.6, 3.6),
col = cols, xlab = "Mean Difference: PMFH - MXF"))
abline(h = 2, lty = 2)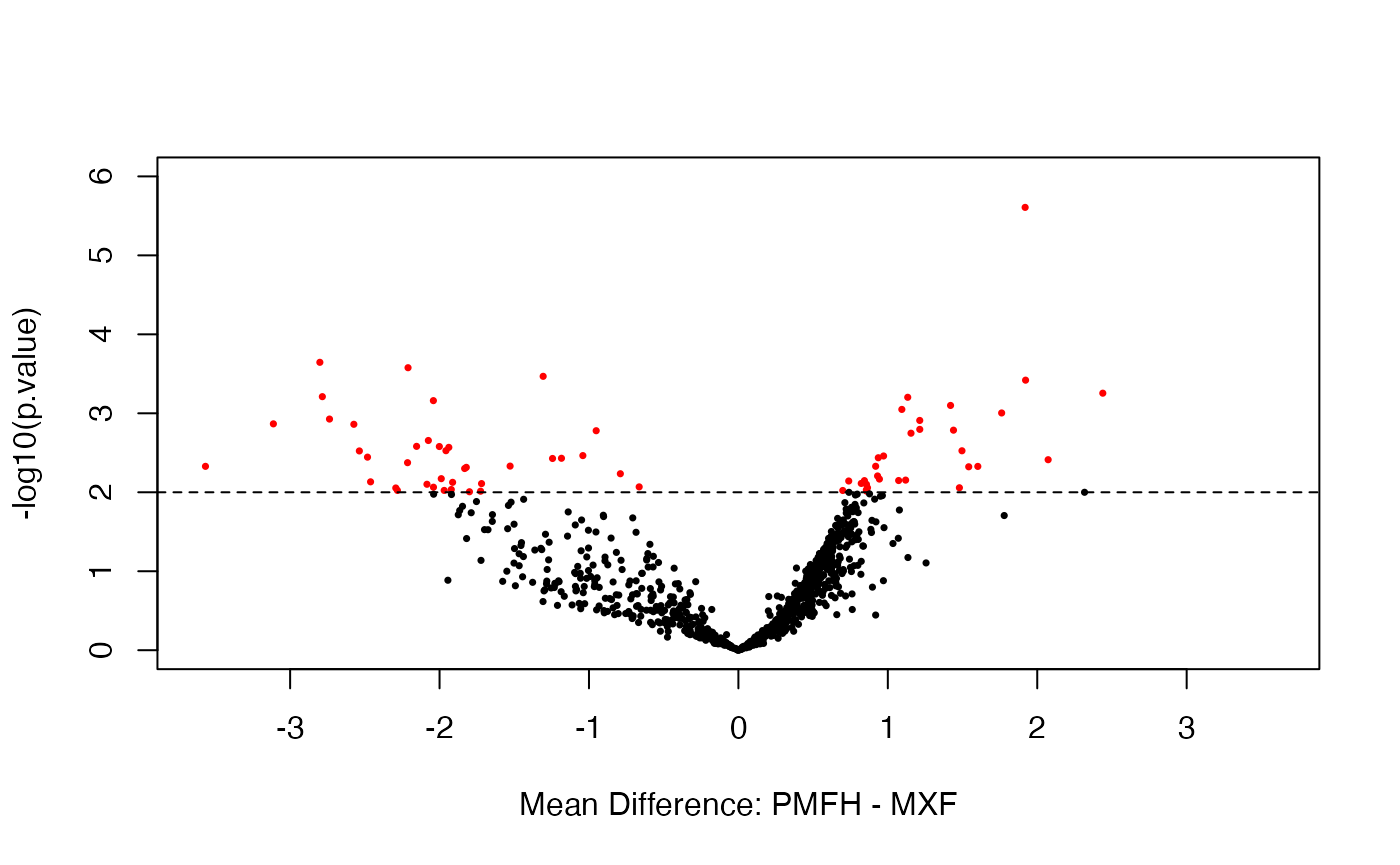
Comparison of Differential Expression Status Between Benchmark Data and Test Data
Significance of differential expression is claimed using a p-value cutoff of 0.01 for both the benchmark data and the test data. They are compared with each other using the Venn diagram.
pval.bench.test <- data.frame(cbind(bench.pval = benchmark.voom$p.val,
test.pval = test.voom$p.val[names(benchmark.voom$p.val)]))
attach(pval.bench.test)
bench.sig <- (bench.pval < 0.01)
test.sig <- (test.pval < 0.01)
venn2 <- cbind(bench.sig, test.sig)
vennDiagram(vennCounts(venn2),
names = c("Benchmark", "Test"),
cex = 1.5, counts.col = rainbow(1))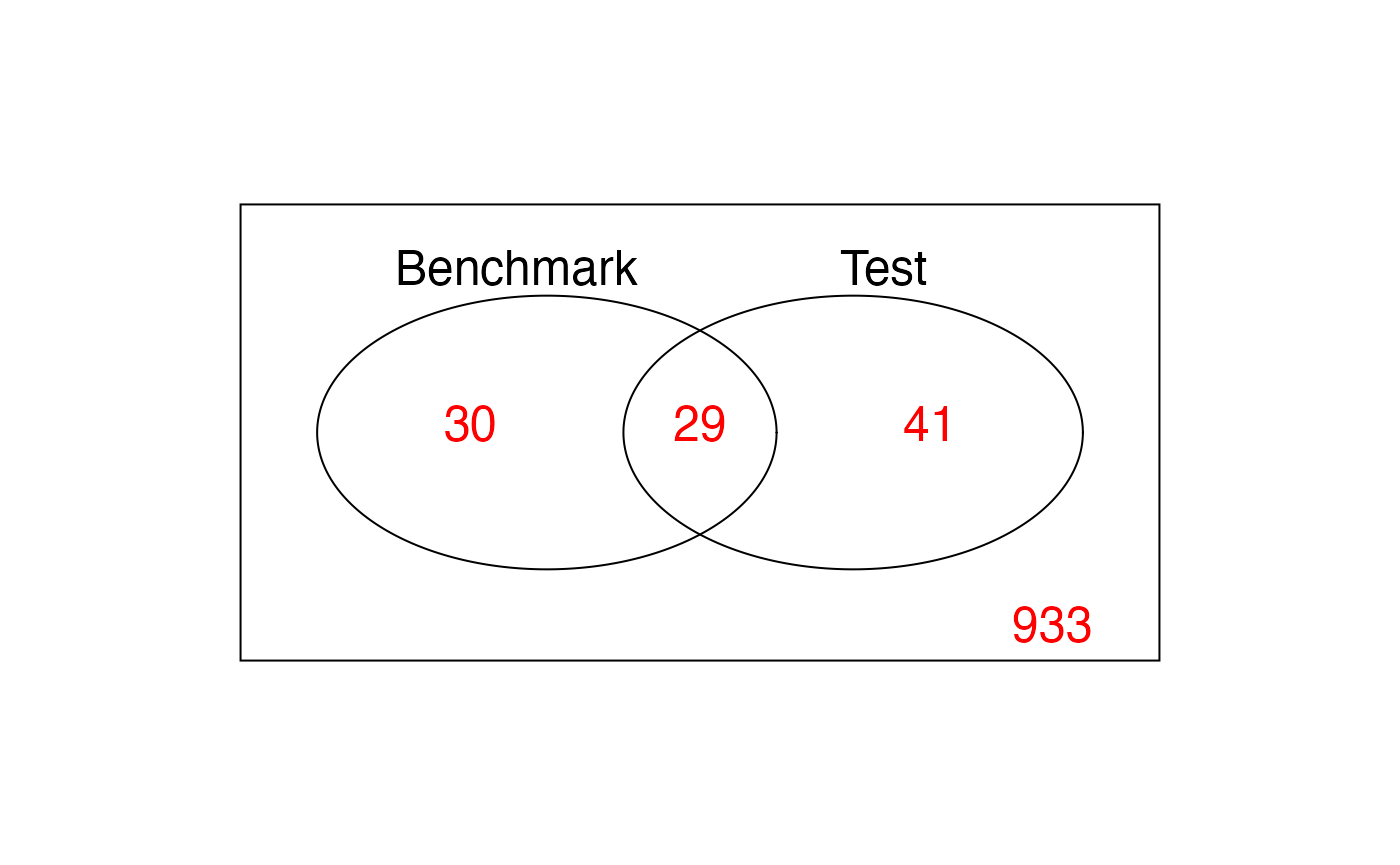
Comparison of Estimated Group Means Between Benchmark Data and Test Data
Scatterplot for the estimated group means for MXF and PMFH between benchmark data and test data.
benchmark.log.MXF.mean <- rowMeans(benchmark.log[, grepl("MXF", colnames(benchmark.log))])
plot(benchmark.log.MXF.mean,
rowMeans(test.log[,grepl("MXF", colnames(test.log))]), pch = 16, cex = 0.5,
xlab = "Group Mean Average of Log2 Count in Benchmark",
ylab = "Group Mean Average of Log2 Count in Test",
main = "MXF", xlim = c(0, 20), ylim = c(0, 20))
abline(0,1)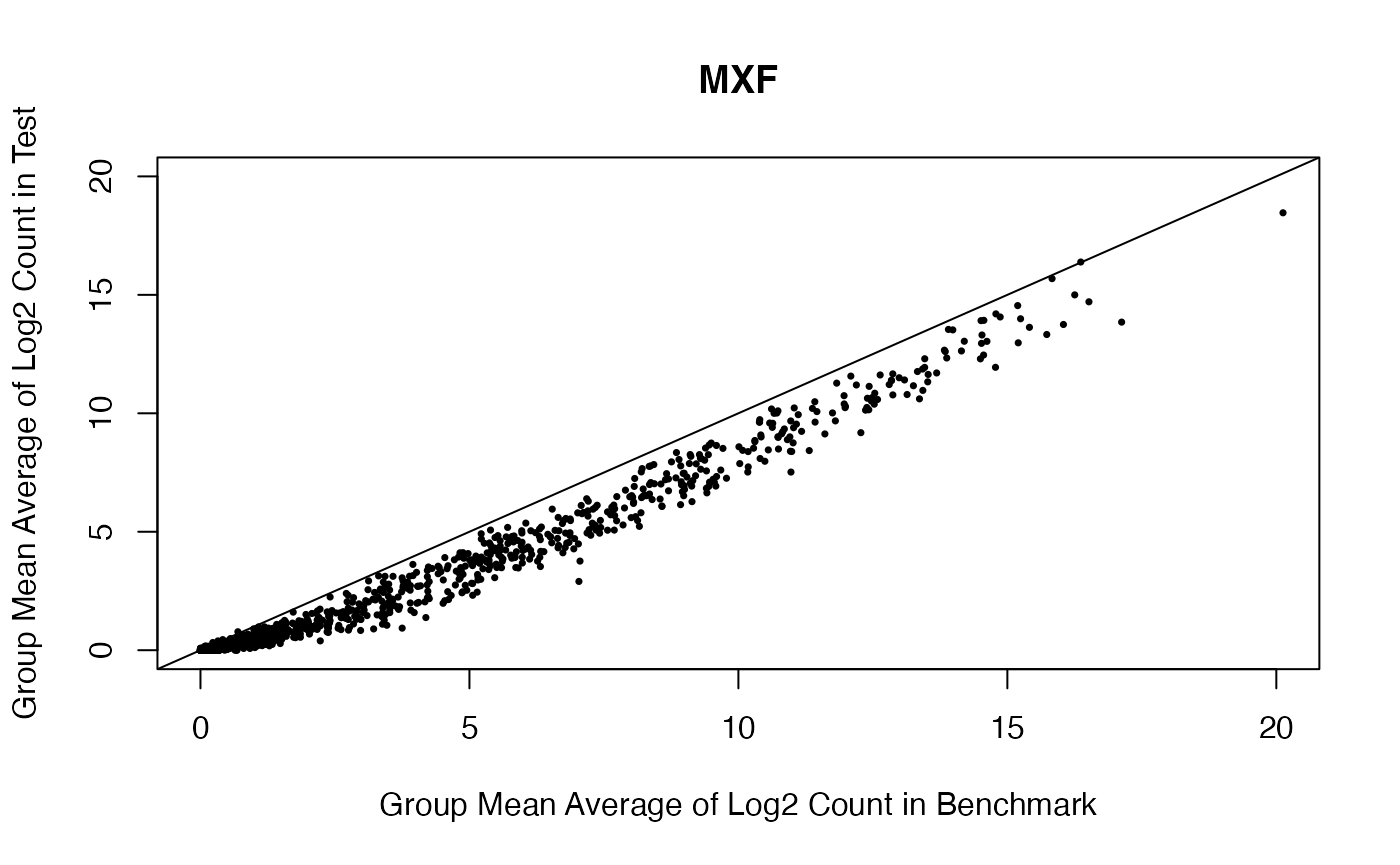
benchmark.log.PMFH.mean <- rowMeans(benchmark.log[, grepl("PMFH", colnames(benchmark.log))])
plot(benchmark.log.PMFH.mean,
rowMeans(test.log[,grepl("PMFH", colnames(test.log))]), pch = 16, cex = 0.5,
xlab = "Group Mean Average of Log2 Count in Benchmark",
ylab = "Group Mean AVerage of Log2 Count in Test",
main = "PMFH", xlim = c(0, 20), ylim = c(0, 20))
abline(0,1)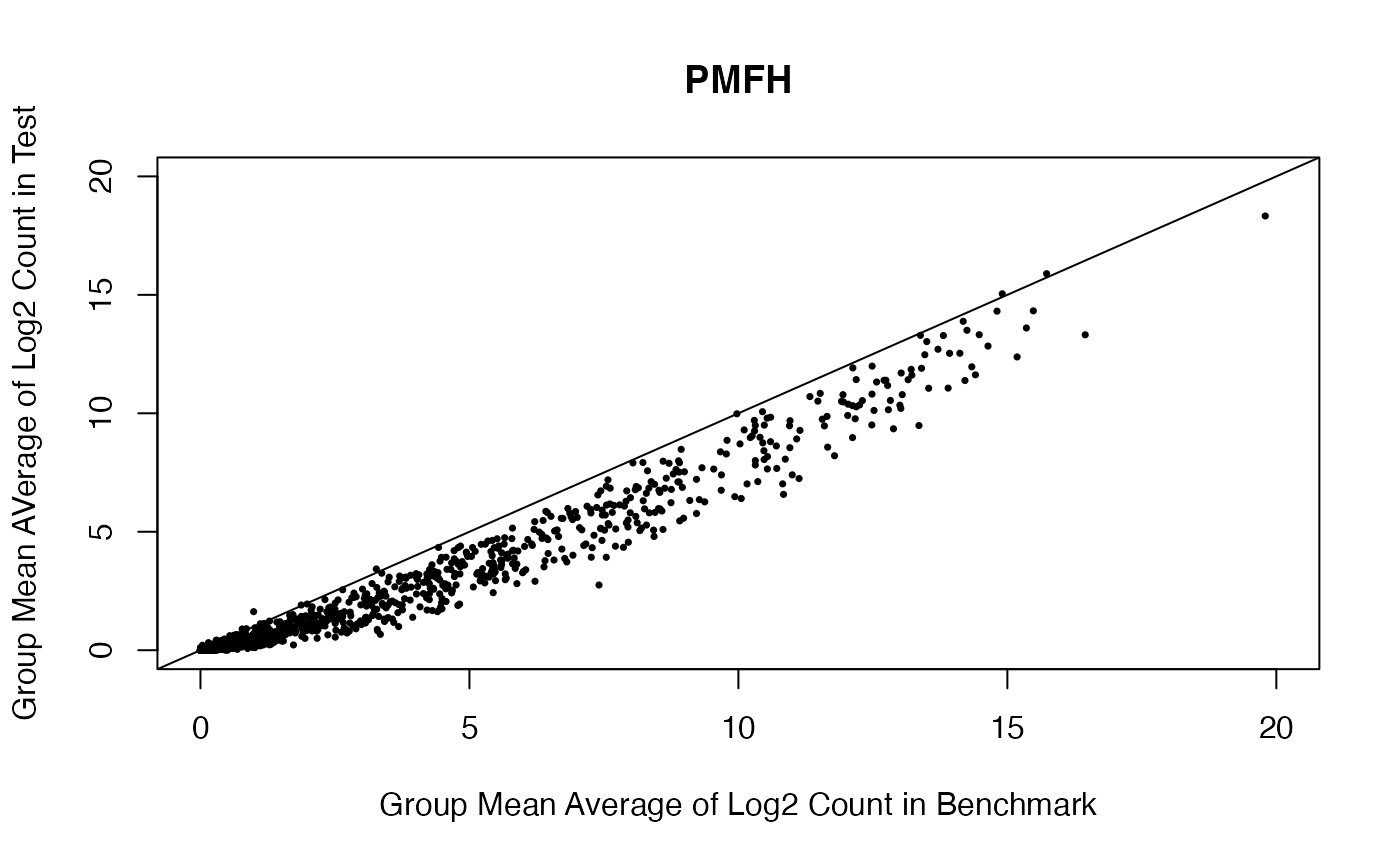
Simulation
We use simulated.data() function to generate additional pairs of data sets by permuting the paired data sets to achieve a pre-specified proportion and magnitude of differential expression. As an example, we show how to generate data sets with the proportion of differential expression around 0.02 (more specifically, in the range of 0.0175 and 0.0225) and the median of mean differences (for log2 counts) around 0 (more specifically, in the range of -0.5 and 0.5).
simulated <- simulated.data(proportion = c(0.0175, 0.0225), median = c(-0.5, 0.5), numsets = 100)
head(simulated[[1]]$simulated_benchmark)[,1:5]
#> D17 D7 D19 E3 E5
#> hsa-let-7a-2* 4 2 2 5 11
#> hsa-let-7a(3) 61589 30934 40506 69704 268842
#> hsa-let-7a*(2) 436 216 196 353 1517
#> hsa-let-7b 29023 8395 25772 17920 77765
#> hsa-let-7b* 125 50 106 92 607
#> hsa-let-7c 9774 5633 17112 13838 65477
head(simulated[[1]]$simulated_test)[,1:5]
#> D17 D7 D19 E3 E5
#> hsa-let-7a-2* 1 2 0 0 2
#> hsa-let-7a(3) 53958 89726 60273 21107 253696
#> hsa-let-7a*(2) 113 368 157 137 1162
#> hsa-let-7b 15832 17546 25624 4744 93419
#> hsa-let-7b* 32 48 73 15 258
#> hsa-let-7c 8156 11921 16656 4445 71733