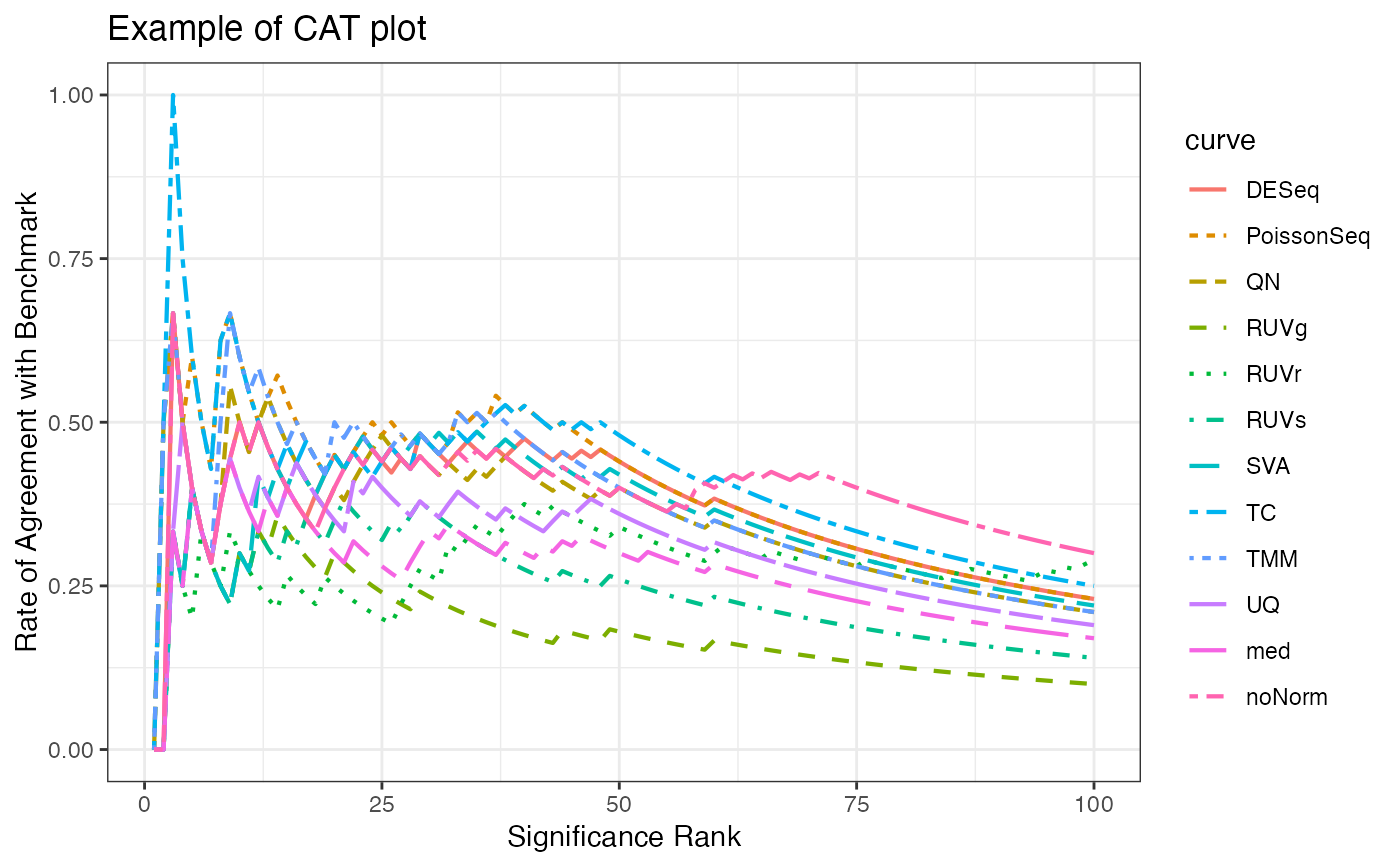
Concordance At The Top Plot
fig.CAT.RdFunction for generating a concordance at the top plot, which compares
concordance of the p-values of differential expression --- typically from the
normalized or raw data set data.test --- to the p-values of
differential expression of a gold standard --- typically
the data set data.benchmark.
fig.CAT(DEA, truth.DEA, title, maxrank = 100, subset = NULL)Arguments
| DEA | Results of a differential expression analysis using the
included methods
|
|---|---|
| truth.DEA | Gold standard (assumed truth) for differential expression.
Must be in the same format as |
| title | optional Figure title |
| maxrank | optional specify the maximum size of top-ranked items that you want to plot. |
| subset | optional vector of a subset of genes/markers for this analysis |
Value
figure of concordance for comparison
Examples
voom.benchmark <- DE.voom(data.benchmark, data.group)
test.norm <- pip.norm(raw=data.test, groups=data.group, norm.method = "all")
#> converting counts to integer mode
#> Loading required package: Biobase
#> Loading required package: BiocGenerics
#> Loading required package: parallel
#>
#> Attaching package: ‘BiocGenerics’
#> The following objects are masked from ‘package:parallel’:
#>
#> clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
#> clusterExport, clusterMap, parApply, parCapply, parLapply,
#> parLapplyLB, parRapply, parSapply, parSapplyLB
#> The following objects are masked from ‘package:stats’:
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from ‘package:base’:
#>
#> Filter, Find, Map, Position, Reduce, anyDuplicated, append,
#> as.data.frame, basename, cbind, colnames, dirname, do.call,
#> duplicated, eval, evalq, get, grep, grepl, intersect, is.unsorted,
#> lapply, mapply, match, mget, order, paste, pmax, pmax.int, pmin,
#> pmin.int, rank, rbind, rownames, sapply, setdiff, sort, table,
#> tapply, union, unique, unsplit, which.max, which.min
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
test.DE <- list(
TMM = DE.voom(RC=test.norm$TMM$dat.normed, groups = data.group),
TC = DE.voom(RC=test.norm$TC$dat.normed, groups = data.group),
UQ = DE.voom(RC=test.norm$UQ$dat.normed, groups = data.group),
med = DE.voom(RC=test.norm$med$dat.normed, groups = data.group),
DESeq = DE.voom(RC=test.norm$DESeq$dat.normed, groups = data.group),
PoissonSeq = DE.voom(RC=test.norm$PoissonSeq$dat.normed, groups = data.group),
QN = DE.voom(RC=test.norm$QN$dat.normed, groups = data.group),
RUVg = DE.voom(RC=data.test, groups = data.group, normalized=FALSE, adjust=test.norm$RUVg$adjust.factor),
RUVs = DE.voom(RC=data.test, groups = data.group, normalized=FALSE, adjust=test.norm$RUVs$adjust.factor),
RUVr = DE.voom(RC=data.test, groups = data.group, normalized=FALSE, adjust=test.norm$RUVr$adjust.factor),
SVA = DE.voom(RC=data.test, groups = data.group, normalized=FALSE, adjust=test.norm$SVA$adjust.factor),
noNorm = DE.voom(RC=data.test, groups = data.group))
fig.CAT(DEA = test.DE, truth.DEA = voom.benchmark, title = "Example of CAT plot")